Today, I want to study the growth plate and show how it is in many ways just a collection of independently differentiating chondrocytes. Remember, the limb lengthening procedure just allows the bodies natural growth to happen by enabling the release of periosteal progeintor cells and the bone marrow within the intermedullary cavity. Our goal with LSJL is to achieve independent differentiation of stem cells into chondrocytes.
A growth plate(source: eMedicine)
The prospective zones from bottom to top are the ossification zone, the calcified cartilage zone, the hypertrophic bone, the proliferative zone, and the rest zone(where stem cells are but there are stem cells in other places as well). Really though you can see the overlap of several zones indicating that chondrocytes are remarkably independent.
The above picture is the periosteum to compare to the growth plate. The cortical bone is the solid red zone. To build new bone, osteoblasts simply deposit new bone according to the OPG/RANKL gradient. The fundamental reason why bone deposits beneath the periosteum and not the longitudinal ends of the bones is that they are not ordered to by OPG. The ossification of cartilage provides a signal to deposit new bone. Distraction Osteogenesis also provides this signal. Articular cartilage is at the end of long bones and the reason it doesn't ossify in normal individuals is lack of CD44 and because there's no OPG telling it to.
Here's a larger photo of a growth plate(source: Wikipedia). It may look like a gap but really the two bones are interconnected:
It is possible to grow taller without the aid of a growth plate like in the picture below which illustrates healing by gap fracture.
source:www.teambone.com
Now this is a cortical bone fracture. Notice the gap between the two spaces of bone. That gap will not go through the process of cartilage to trabecular bone etc. and eventually become cortical bone(only larger fractures will undergo that process). If that gap occurs horizontally it will result in a slightly taller individual. Over time that could result in a very large height gain. Now if you get microfractures on a smaller scale those tiny little gaps will eventually heal resulting in height gain(albeit an unequal height gain).
I don't think trying to increase chondrocyte proliferation in pre-fused growth plate teens is the way to go(Chondrocytes have to have their proliferative capacity adjusted first). However, the faster chondrocytes proliferate the faster you will grow taller.
"The growth of fetal and postnatal bones [was]stimulated by IGF1 (100 ng/ml) and inhibited by dexamethasone (Dexa; 1 microM). We found that the capacity to undergo catch-up growth was restricted to postnatal bones. Catch-up growth occurred after postnatal bones had been exposed to Dexa for 7 or 12 days but not after a more prolonged exposure (19 days). Incomplete catch-up growth resulted in compromised bone length when assessed at the end of the 4-month period of culture. While exposure to Dexa was associated with decreased chondrocyte proliferation and differentiation, catch-up growth was only associated with increased cell proliferation. The phenomenon of catch-up growth after Dexa treatment is intrinsic to the growth plate and primarily mediated by an upregulation of chondrocyte proliferation."
"The direct effects of Dexa are believed to be partly caused by downregulation of the GH receptor and/or the IGF1 receptor at the growth plate level"
GHR and IGF1R are very important as they determine the growth plate response to GH and IGF-1. GHR and IGF1R are more important than GH and IGF-1. Dexamethasone can induce chondrogenic differentiation in stem cells which is why it is so appealing. But it seems that the growth plate can not recover from the loss of GH and IGF-1 receptors. Therefore, a way to increase chondrocyte proliferation is to not take Dexamethasone.
The growth plate has the ability to downregulate and upregulate chondrocyte proliferation. Any method that increases chondrocyte proliferation has to get around this positive and negative feedback mechanism.
Postnatal refers to phenomenon occurring after birth.
"Ihh expression in postnatal chondrocytes has a non-redundant role in maintaining a growth plate and sustaining trabecular bone after birth. Loss of Ihh in postnatal chondrocytes results in fusion of the growth plate and a decrease in trabecular bone....Expression of a Jansen receptor in chondrocytes was able to rescue abnormal chondrocyte differentiation but not impaired chondrocyte proliferation and the bone anomalies in mice lacking the Ihh gene in chondrocytes after birth. Taken together, our findings suggest that Ihh has both PTHrP-dependent and -independent functions during postnatal endochondral bone development."
"The Ihh signal is transduced through smoothened (Smo), a putative G protein-coupled seven-transmembrane domain protein. In the absence of Ihh protein, Smo is repressed by the Ihh target gene patched (Ptch), another cell surface receptor for Ihh."
"overexpression of either Ihh or a constitutively active Smo allele specifically in cartilage results in increased activity of the Ihh signaling pathway to promote chondrocyte proliferation". Ihh also induces Cyclin D1.
"collagen type 2 is also expressed in the kidney"<-maybe we can recruit COL2A1 from there?
Chondrocyte differentiation refers to mesenchymal stem cells turning into chondrocytes usually in cartilage. Osteoblast differentiation refers to the ability for mesenchymal stem cells to turn into osteoblasts usually in bone.
If you're a teenager and your indian hedgehog isn't working then that's some 'free growth' that has potentially been lost as Ihh is an important factor in chondrocyte proliferation.
Chondrocyte p21(WAF1/CIP1) expression is increased by dexamethasone but does not contribute to dexamethasone-induced growth retardation in vivo.
"The inhibitory effects of glucocorticoids (GCs) on chondrocyte proliferation are consistent with GCs disrupting cell cycle progression and promoting cell cycle exit. Cyclin-dependent kinase inhibitors (CDKIs) force cells to exit the cell cycle and differentiate, and studies have shown that expression of the CDKI p21(CIP1/WAF1) is increased in terminally differentiated cells. p21 mRNA and protein expression was increased during chondrocyte differentiation and after exposure to dexamethasone (Dex, 10(-6 )M) in murine chondrogenic ATDC5 cells... In addition, p21 ablation had no effect on the reduction in width of the growth plate or reduced mineral apposition rate in Dex-treated mice. However, an alteration in growth rate and epiphyseal structure is evident when comparing p21(-/-) and wild-type mice. p21is involved in the maintenance of the growth plate."
"In mammals, there are two families of CDKIs: the CIP/KIP family, composed of p21WAF1/CIP1, p27KIP1, and p57KIP1, which bind to and inhibit G1 CDKs, and the INK4 proteins p16INK4A, p15INK4B, p18INK4C and p19INK4D, which inhibit CDK4 and, to a lesser extent, CDK6. The Cip/Kip family acts on most cyclin/CDK complexes and is essential to inhibit G1 progression and S1 entry"
"Disruption of the p57 gene has been shown to cause delayed chondrocyte differentiation, resulting in skeletal deformations and shortened limbs"<-maybe increasing p57 expression can increase height?
"mice deficient in p27 [are larger]"
"In chondrocytes, p21 expression is p38/MAPK-dependent"
"p21 blocks the formation of cyclin/CDK complexes, which promote cell cycle progression through the phosphorylation of pocket proteins pRb (retinoblastoma protein), p107, and p130. Once phosphorylated, these pocket proteins form complexes with E2F transcription factors, leading to the transcription of genes necessary for cell cycle progression and DNA replication."
p21 could be very valuable. P21 could be vital in the formation of new growth plates.
"Chondrogenesis and subsequent endochondral ossification are processes tightly regulated by the transcription factor Sox9 (SRY-related high mobility group-Box gene 9). coactivator-associated arginine methyltransferase 1 (CARM1) regulates chondrocyte proliferation via arginine methylation of Sox9. CARM1-null mice display delayed endochondral ossification and decreased chondrocyte proliferation. Conversely, cartilage development of CARM1 transgenic mice was accelerated. CARM1 specifically methylates Sox9 at its HMG domain in vivo and in vitro. Arg-methylation of Sox9 by CARM1 disrupts interaction of Sox9 with beta-catenin, regulating Cyclin D1 expression and cell cycle progression of chondrocytes. CARM1 [is] an important regulator of chondrocyte proliferation during embryogenesis." "CARM1 has been shown to synergistically activate transcription with nuclear receptors in combination with other coactivators, such as p160 family, p300/CBP and SRC-2/TIF2/GRIP1. After recruitment to promoters of steroid-responsive genes, CARM1 methylates specific residues (Arg17 and Arg26) at the N-terminus of histone H3 resulting in transcriptional activation"
CARM1 null mice had reduced height. CARM1 transgenic mice had increased height.
"Sox9 could be methylated by CARM1/PRMT4"
"Decreased chondrocyte proliferation phenotypes seen in Sox9 transgenic mice are partly explained by the model that Sox9 competes with Tcf/Lef for binding to beta-catenin and regulates chondocyte proliferation via Cyclin D1 expression. The beta-catenin/Tcf complex binds the Tcf/Lef consensus site in the Cyclin D1 promoter, transactivating Cyclin D1" Sox9 methylation by CARM1 inhibits Sox9 degradation of Beta-Catenin.
"CARM1 and Sox9 may cooperatively regulate CyclinD1 promoter activity."
So CARM1 can increase height and allow for more Sox9 by inhibiting the likely mechanism where too much Sox9 decreases height by inhibiting Cyclin D1.
"The longitudinal growth of bones is apparently controlled by modifying the numbers of growth plate chondrocytes in the proliferative zone, their rate of proliferation, the amount of chondrocytic hypertrophy and the controlled synthesis and degradation of matrix throughout the growth plate. These variables may be modulated to produce a change in growth rate in the presence of sustained or cyclic mechanical load. Tissue and cellular deformations involved in the transduction of mechanical stimuli depend on the growth plate tissue material properties that are highly anisotropic, time-dependent, and that differ in different zones of the growth plate and with developmental stages. There is little information about the effects of time-varying changes in volume, water content, osmolarity of matrix, etc. on differentiation, maturation and metabolic activity of chondrocytes. Also, the effects of shear forces and torsion on the growth plate are incompletely characterized. Future work on growth plate mechanobiology should distinguish between changes in the regulation of bone growth resulting from different processes, such as direct stimulation of the cell nuclei, physico-chemical stimuli, mechanical degradation of matrix or cellular components and possible alterations of local blood supply." "The shapes of joint profiles may be determined in part by mechanical loading"
"Chondrocytic enlargement and matrix synthesis are strongly correlated since chondrocytes enlarge mostly in the growth direction, with little increase in their width. Hence matrix synthesis is required to fill the increased volume laterally surrounding each cell as it grows in the longitudinal direction"
"Growth plate mechanical behavior is comparable to articular cartilage"
"It is hypothesized that the more rigid reserve zone, which also constitutes a greater proportion of growth plate height in large animals, might provide mechanical support in growth plates of large species that are undergoing slower growth rates for long periods of time together with greater mechanical loading"
"There is a significant increase in the extracellular matrix elastic moduli from the reserve zone to the chondro-osseous junction"
"The length of the playing-side ulnar and 2nd metacarpal were respectively 3% and 3.7% longer than on the contralateral side [in adult tennis players]"
Altering the controlled synthesis or degradation of matrix may be a way to increase height growth. It's interesting that the study mentions that the rate of chondrocyte proliferation may affect height growth. Maybe the rate of chondrocyte proliferation could affect growth by upregulating certain hormones or by changing the structure of the bone(more proliferating chondrocytes = larger growth plate).
Mesenchymal Stem Cells may be another method of encouraging chondrocyte proliferation. Trophic effects of mesenchymal stem cells increase chondrocyte proliferation and matrix formation.
"coculture of primary chondrocytes (PCs) with various sources of multipotent cells results in a higher relative amount of cartilage matrix formation than cultures containing only chondrocytes[if you add MSCs to a cartilagenous matrix then more ECM will be deposited]. We used coculture pellet models of human mesenchymal stem cells (hMSCs) and human PCs or bovine PCs (bPCs) and studied the fate and the contribution to cartilage formation of the individual cell populations during coculture. Enhanced cartilage matrix deposition was confirmed by histology and quantification of total glycosaminoglycan deposition. Species-specific quantitative polymerase chain reaction demonstrated that cartilage matrix gene expression was mainly from bovine origin when bPCs were used. Short tandem repeat analysis and species-specific quantitative polymerase chain reaction analysis of genomic DNA demonstrated the near-complete loss of MSCs in coculture pellets after 4 weeks of culture. In coculture pellets of immortalized MSCs and bPCs, chondrocyte proliferation was increased, which was partly mimicked using conditioned medium, and simultaneously preferential apoptosis of immortalized MSCs was induced. Taken together, our data clearly demonstrate that in pellet cocultures of MSCs and PCs, the former cells disappear over time. Increased cartilage formation in these cocultures is mainly due to a trophic role of the MSCs in stimulating chondrocyte proliferation and matrix deposition by chondrocytes rather than MSCs actively undergoing chondrogenic differentiation[this is very interesting, maybe the limit in chondrocyte proliferative capacity applies to MSCs too so injecting new MSCs into the growth plate would not exceed this capacity]."
If new growth plate MSCs don't exceed proliferative capacity by a feedback mechanism within the chondrocytes then LSJL may be less effective in open growth plates. There is ample evidence that MSCs can differentiate into chondrocytes under hydrostatic pressure. With active plates, LSJL may only increase matrix formation whereas with no cartilagenous growth plate LSJL doesn't have to worry about negative feedback from "Primary chondrocytes".
"evidence suggests that differentiation into tissue-specific cells cannot fully explain the benefits of transplanted MSCs in remodeling and recovery of damaged or lost tissue"<-So MSCs may mostly encourage existing cell activity.
"it has been suggested that increased cartilage formation in cocultures is due to chondrogenic differentiation of MSCs stimulated by factors secreted by chondrocytes. Indeed, chondrocyte-conditioned medium can induce chondrogenic differentiation of MSCs directly and in transwell cultures[a mechanism like LSJL]"
"DNA analysis of coculture pellets at 1, 2, and 3 weeks of culture demonstrated a steep drop in human DNA between 1 and 2 weeks with a further gradual decline at weeks 3 and 4"<-So the amount stem cell DNA decreased which you would not expect if stem cells were differentiating into chondrocytes.
"Even in cell pellets with an initial seeding of 80% iMSCs, hardly any human mRNA was detected"
"Cell death in iMSC-containing pellets started to increase significantly from day 5 onward."
"These data show that the change in ratio between MSC and PC during prolonged coculturing is in addition to apoptosis also due to increased proliferation of chondrocytes in pellet cultures."<-So maybe MSCs do help exceed proliferative capacity just not by proliferating themselves.
"By providing nutrients and growth factors, MSCs increase proliferation and differentiation of host-derived cells to help them to repair damaged tissues."<-So when there are already enough cells, MSCs provide resources. However whether this increases proliferative capacity is unclear.
The same process of endochondral ossification is used to both naturally increase height and to heal fractures. It is also used to heal microfractures dependent on the type of microfracture it is. Chondrocytes are an important part of endochondral ossification and can result in height increase as chondrocytes can reproduce dependent on telomere length and DNA Methylation status. Chemical signaling mechanisms like Growth Hormone have failed to reproduce Gigantism likely due to these two factors. However, there still has to be a reason that Gigantism exists. Still, chemical and dietary means of increasing chondrocyte proliferation can result in faster healing of fractures plus faster endochondral ossification and therefore faster increasing of height. Effect of Nell-1 Delivery on Chondrocyte Proliferation and Cartilaginous Extracellular Matrix Deposition "Cartilage tissue engineering using chondrogenic growth factors is an attractive strategy to promote cartilage repair. Bone morphogenetic proteins have been widely studied for their application in cartilage repair. However, functional heterogeneity of bone morphogenetic proteins and unpredictable effects such as cyst formation may limit their therapeutic use. Thus, the use of alternative growth factors with greater osteochondral specificity may be advantageous for cartilage regeneration. Nel-like molecule-1 (Nell-1; Nel is a protein strongly expressed in neural tissue encoding epidermal growth factor-like domain) is a novel growth factor believed to specifically target cells committed to the osteochondral lineage. Mutation of the Nell-1 gene has been shown to disrupt normal cartilage growth and development in rodents. This study investigates the chondrogenic potential of recombinant human Nell-1 protein in a three-dimensional alginate hydrogel microenvironment containing rabbit chondrocytes. To provide controlled delivery and maximize biological efficiency, Nell-1 was incorporated in chitosan microparticles. Over 42 days of culture, chondrocyte proliferation and cluster formation was significantly enhanced by Nell-1 in a dose-dependent manner. Further, the clusters formed in the presence of Nell-1 contained more type II collagen and glycosaminoglycans than clusters formed within Nell-free control gels. These findings demonstrate the ability of Nell-1 to promote chondrocyte proliferation and deposition of cartilage-specific extracellular matrix materials." Currently, there are no food or supplements that contain Nell-1 but it is patented so one can assume that future products will appear on the horizon(given how many things are turned into supplements). Nell-1 definitely increases growth rate but whether Nell-1 will actually increase height is unknown. Mechanisms of impaired growth: effect of steroids on bone and cartilage. "Systemically, GC(glutocorticoids-an example is cortisol) treatment reduces circulating levels of estrogen and modestly increases parathyroid hormone levels. At the local level, GCs decrease insulin-like growth factor I (IGF-I) production, induce IGF-I resistance and increase nuclear factor kappaB ligand(also known as RANKL) production by osteoblasts. These alterations inhibit new bone formation and stimulate bone resorption, with a net loss of bone over time. Clinically, this results in decreased bone mineral density, osteoporosis and increased risk for fracture. Local effects of GCs at the growth plate include reduction of IGF-I production, inducing IGF-I resistance and reducing production of C-type natriuretic peptide[the C-type natruiretic peptide is very important in height growth, overexpression of CNP has resulted in enhanced height growth], which results in a reduction of chondrocyte proliferation, matrix synthesis and hypertrophy. These reductions in chondrocyte function result in decreased linear growth." Glutocorticoids are a specific type of steroid. We know that bodybuilders are always trying to find ways to lower their cortisol. It's unclear whether there's catch up production of CNP in response to say increased cortisol levels.
SCF, BDNF, and Gas6 are regulators of growth plate chondrocyte proliferation and differentiation. "Bovine epiphyseal chondrocytes separated by density gradient centrifugation differ in proliferative response to IGF-I and IGF-I receptor number. To identify novel modifiers of IGF-I action at the growth plate, we used microarray analyses to compare bovine hypertrophic and reserve zones and identified several receptors differentially expressed across the growth plate: NTRK2 [receptor for brain-derived neurotrophic factor (BDNF)], KIT [receptor for stem cell factor (SCF)], and MER and AXL [two receptors for growth arrest-specific 6 (Gas6)]. The corresponding ligands were tested for their ability to stimulate either proliferation of isolated chondrocytes or differentiation in ATDC5 cells. Each factor inhibited IGF-I-mediated proliferation in isolated chondrocytes by attenuating ERK1/2 activation[so NTRK2, KIT, MER, and AXL dampen ERK1/2 activation].SCF, BDNF, Gas6, and C-type natriuretic peptide promoted differentiation in ATDC5 cells[Since CNP can increase height maybe SCF, BDNF, and Gas6 can increase height as well], each factor producing different expression patterns for collagen X, collagen 2, aggrecan, and lysyl oxidase. Whereas multiple factors stimulated ATDC5 differentiation, only IGF-I and high-dose insulin, out of several factors implicated in chondrocyte maturation, stimulated proliferation of isolated chondrocytes. IGF-I appears to be the primary proliferative signal in growth plate chondrocytes, whereas multiple factors including SCF, BDNF, and Gas6 regulate the pace of differentiation at the growth plate." IGF-1 is what primarily increases chondrocyte proliferation but SCF, BDNF, Gas6, and CNP may increase height at other stages of growth.
Both Chondroinduction and Proliferation Account for Growth of Cartilage Nodules in Mouse Limb Bud Cultures.
"High density micromass culture of limb bud mesenchymal stem cells isolated from mouse embryos represents a well-established model to study chondro- and osteogenesis. To determine whether cartilage nodules grow solely by induction of surrounding cells or proliferation of cells within the nodules, we performed BrdU/Collagen II (Col II) double-labelling and 3D reconstruction of growing cartilage nodules. Col II-positive replicating chondrocytes are present throughout the nodules with the majority of replicating cells localized on the top (cell-medium interface) and periphery/sides of nodules. Kinetic analysis of cellular proliferation within the nodules demonstrated the time-dependent reduction in number of Col II-positive replicating cells. The sequential expression of Col I, Col II, Col X, parathyroid hormone related peptide receptor 1 (Pthr1), bone sialoprotein (Bsp) and osteocalcin (Ocn) mRNAs was similar to that characterizing chondrocyte differentiation and maturation in vivo. We conclude that the limb bud model recapitulates events seen during endochondral bone formation: cellular aggregation, proliferation, differentiation and maturation to hypertrophy. We also conclude that not only induction of peri-nodular mesenchymal cells but also proliferation of chondrocytes within cartilage nodules contribute to cartilage nodule growth."
Decrease in chondrocyte proliferation(inferred from decrease in Type II Collagen) occurred before decrease in chondrocyte hypertrophy meaning that perhaps maintaining chondrocyte proliferation can ward off growth cessation.
This study to be analyzed in more detail in the future.
Previously, running was analyzed in it's ability to induce bone adaptation or cause microfractures. Now an equally high impact exercise known as boxing will be analyzed.
Boxer's fracture
"Boxer's fracture is a common name for a fracture of the distal fifth metacarpal and received its name from one of its most common causes, punching an object with a closed fist. It can occur from a fistfight or from punching a hard object."
Repeated fractures and healing of the metacarpals would result in an increased hand size. Does anyone know of any instances observed of this?
[Modification of bone quality by extreme physical stress. Bone density measurements in high-performance athletes using dual-energy x-ray absorptiometry]
"Little is known on the influence of high performance sports on the bone density especially in athletes with high demands on weight bearing of the spine. In (n = 40) internationally top ranked high performance athletes of different disciplines (n = 28 weight-lifters, n = 6 sports-boxers and n = 6 bicycle-racers) bone density measurements of the lumbar spine and the left hip were performed. The measurements were carried out by dual-photonabsorptiometry (DEXA; QDR 2000, Siemens) and evaluated by an interactive software-programme (Hologic Inc.). The results were compared to the measurements of 21 age-matched male control individuals. In the high performance weight lifters there was an increase of bone density compared to the control individuals of 23% in the Ward's triangle (p < 0.01). The sports-boxers had an increase up to 17% (lumbar spine), 9% (hip) and 7% (Wards' triangle). In the third athletes group (Tour de France-bikers) BMD was decreased 10% in the lumbar spine, 14% in the hip and 17% in the Wards' triangle."
Remember DEXA does not measure volumetric BMD but aereal BMD; and aereal BMD incorporates size of the bone. The ward's triangle is in the neck part of the femur bone. Elite boxers do a variety of training so you can't really determine what causes what(confounding variables). Weight lifters could have naturally larger bones so this study can't really be used to detect microfractural causes. .Boxing causes a lot of spiral forces on the spine due to the twisting motion that occurs during a well executed punch. It's interesting that tour de France-bikers had a reduced bone density versus controls. This could indicate that lower bone density is beneficial for cycling as a sport. We don't know exactly what caused the increase in aereal bone density but it could be something that increases height like something involving the periosteum or new cortical bone deposition.
Hand injuries in boxing.
"Investigations of 100 consecutive hand injuries in boxing have shown that 39% occurred in the area of the thumb, including the radial carpals, metacarpals and phalanges and their joints. These injuries were mostly caused by forced abduction of the thumb. Thirty-five percent of the injuries occurred at the base of Metacarpals 2 to 5, including the wrist joint, and arose from forced flexion of the wrist. Twenty-six percent occurred in the phalanges and the rest of the metacarpals, excluding the bases."
If you're trying to cause tensile strain microfractures for instance. Forced flexion may be a way to induce them. Maybe stretching is a plausible way to induce forced flexion on the bone thus causing tensile strain microfractures.
There's not nearly as much research on boxing fractures as their is on running fractures. In any sport there are always confounding variables because most athletes incorporate a variety of training methods. However, bag punching remains a possible viable way to create microfractures in the hand and wrist. Punching seems superior to kicking as you can generate more impact relative to the strength of the bone in punching versus kicking.
The metacarpals are short bones so maybe only increasing periosteal width is needed(would have to see the histology to be sure as there could be periosteal patches) and it would be logical to assume that heavy bag punching would cause shearing forces on those bones. As for the fingers, they can be increased in size via lateral synovial joint loading. You can get a lot of leverage on your fingers so all you have to is pinch at the synovial joints of your fingers to result in an increase in bone length.
Boxing did beat weight lifting in increasing bone density in the spine. Boxing causes a lot of shearing forces like by punch rotation. So boxing should have very similar benefits to that achieved by interstitial fluid flow. However, the effects of boxing can be mimicked via weight lifting exercise i.e. you can increase hand size by pushing your body off the floor with your hands and then landing on those hands(this would be an example of lateral tensile strain microfractures) and you can perform body rotations with weights mimicking the spiral forces that occur when a trained boxer throws a punch.
Last blog entry, we learned that high impact sprinting can cause an increase in cortical area via microfractures. Now cortical bone has different adaptive mechanisms for length versus width but if the RANKL/OPG axis was altered then there may be a way to encourage bone deposition at the top of cortical bone. Sprinters were actually shorter than sedentary individuals. This could be due to running causing compression microfractures and decreasing height but increasing width over time. Or, it could be just that shorter individuals prefer running.
Much has been made of growth hormone(GH) in attempting to make people taller.
The over 7-feet tall Richard Kiel(actor)
According to (http://www.medstudents.com.br/endoc/endoc8.htm) gigantism has an incidence rate of about "[3 per 1 million people]". It defines gigantism as "chronic exposure to GH."
High dose growth hormone treatment limited to the prepubertal period in young children with idiopathic short stature does not increase adult height.
"40 children with no signs of puberty, age at start 4-8 years (girls) or 4-10 (boys) years, height SDS<2.0 SDS and birth length>-2.0 SDS, were randomly allocated to receive GH at a dose of 2 mg/m2/day (equivalent to 75 mug/kg/day at start and 64 mug/kg/day at stop) until the onset of puberty for at least 2 years (preceded by two 3-month periods of treatment with low or intermediate doses of GH separated by two washout periods of 3 months) or no treatment."
It is possible that the dosage of Growth Hormone was too low.
"In 28 cases adult height (AH) was assessed at a mean (SD) age of 20.4 (2.3) years. Results: GH-treated children (mean treatment period on high dose GH 2.3 yr (range 1.2-5.0 yr)) showed an increased mean height SDS at discontinuation of treatment compared with controls (-1.3 (0.8) SDS versus -2.6 (0.8) SDS, respectively). However, bone maturation was significantly accelerated in the GH-group compared with controls (1.6 (0.4) versus 1.0 (0.2) yr, respectively) and pubertal onset tended to advance. After an untreated interval of 3-12 yr, AH was -2.1 (0.7) and -1.9 (0.6) in the GH-treated and control group, respectively. Age was a positive predictor of adult height gain."
Growth Hormone only accelerated growth. It did not increase overall adult height. The key component of proving an hypothesis is that the experiment has to be reproducable. Their are clearly other components involved in gigantism, perhaps a genetic mutation affecting the epiphysis in addition to a large amount of growth hormone produced like DNA hypermethylation. Gigantism already involves one cancerous tumor in the pituitary gland(and occasionally other places) why not others?
"Patients were randomly and blindly assigned to treatment with either 3 IU (group A) or 6 IU (group B) GH/m(2).d ( approximately 0.033 or 0.067 mg/kg.d, respectively). The mean (+/-SD) birth length was -3.6 (1.4), the age at the start of the study was 8.1 (1.9) yr, and the height SD score (SDS) at the start of the study -3.0 (0.7). Seventeen of the 54 children were partially GH deficient (stimulated GH peak, 10-20 mU/liter). Fifteen non-GH-treated, non-GH-deficient, short children born SGA, with similar inclusion criteria, served as controls [mean (+/-SD) birth length, -3.3 (1.2); age at start, 7.8 (1.7) yr; height SDS at start, -2.6 (0.5)]."
The dosage of growth hormone was less than the previous study.
"GH treatment resulted in an AH above -2 SDS in 85% of the children after a mean (+/-SD) GH treatment period of 7.8 (1.7) yr. The mean (SD) AH SDS was -1.1 (0.7) for group A and -0.9 (0.8) for group B, resulting from a mean (+/-SD) gain in height SDS of 1.8 (0.7) for group A and 2.1 (0.8) for group B. No significant differences between groups A and B were found for AH SDS (mean difference, 0.3 SDS; 95% confidence interval, -0.2, 0.6; P > 0.2) and gain in height SDS (mean difference, 0.3 SDS; 95% confidence interval, -0.1, 0.7; P > 0.1). When corrected for target height, the mean corrected AH SDS was -0.2 (0.8) for group A and -0.4 (0.9) for group B. The mean (+/-SD) AH SDS of the control group [-2.3 (0.7)] was significantly lower than that of the GH-treated group (P < 0.001). Multiple regression analysis indicated the following predictive variables for AH SDS: target height SDS, height SDS, and chronological age minus bone age (years) at the start of the study. GH dose had no significant effect."
Conclusion: Don't waste your time and money on GH. There's another element of gigantism that's missing.
Other possible causes of gigantism(from www.medstudents.com):
"A rare form [of gigantism is] caused by hypersecretion of GHRH from an ectopic source (pancreatic islet or carcinoid tumors) or from within the central nervous system such as ganglyoneuroma (called eutopic). Even more rare form is a nonpituitary GH secretingtumor documented in a few lung tumors and in those called Ectopic Pituitary Tumor (Esphenoidal)."
All of these causes are still related to GH however. It is clear that gigantism is correlated with high volumes of growth hormone. Since long bone growth does not occur after the growth plates have closed in gigantism(except for Robert Wadlow and other cases) the signs point to the fact that individuals with gigantism probably have special characteristics in the epiphyseal part of their bones. It may be worthwhile to study this to try to apply to our pursuits.
Growth Hormone may be related to DNA Methylation which controls growth plate senesence.
DNMT3A and DNMT3B mediate autocrine hGH repression of plakoglobin gene transcription and consequent phenotypic conversion of mammary carcinoma cells
"Directed by microarray analyses, we report that autocrine human growth hormone (hGH) increased the mRNA and protein expression of DNA methyltransferase 1 (DNMT1), DNMT3A and DNMT3B in mammary carcinoma cells. Autocrine hGH stimulation of DNMT3A and DNMT3B expression was mediated by JAK2 and Src kinases, and treatment of mammary carcinoma cells with the DNMT inhibitor, 5′-aza-2′-deoxycytidine (AZA), abrogated autocrine hGH-stimulated cellular proliferation, apoptosis and anchorage-independent growth. AZA reversed the epitheliomesenchymal transition of mammary carcinoma cells induced by autocrine hGH, to an epithelioid morphology and abrogated cell migration stimulated by autocrine hGH. Autocrine hGH-stimulated hypermethylation of the first exon of the PLAKOGLOBIN gene and AZA abrogated the ability of autocrine hGH to repress plakoglobin gene transcription. Small interfering RNA (siRNA)-mediated depletion of the individual DNMT molecules did not release autocrine hGH repression of PLAKOGLOBIN promoter activity nor did individual DNMT depletion affect autocrine hGH-stimulated migration. However, concomitant siRNA-mediated depletion of both DNMT3A and DNMT3B abrogated hypermethylation of the PLAKOGLOBIN gene stimulated by autocrine hGH and subsequent repression of plakoglobin gene transcription and increased cell migration. Thus, the autocrine hGH-stimulated increases in DNMT3A and DNMT3B expression mediate repression of plakoglobin gene transcription by direct hypermethylation of its promoter and consequent phenotypic conversion of mammary carcinoma cells. Autocrine hGH, therefore, utilizes DNA methylation as a mechanism to exert its oncogenic effects in mammary carcinoma cells."
"Plakoglobin is a component of the adherens junctions complex, and is a multifunctional protein involved in cell–cell adhesion and transcriptional regulation. Loss of plakoglobin expression results in dissolution of adherens junctions"
"CpG hypermethylation of the plakoglobin promoter has also been demonstrated to be responsible for the loss of plakoglobin expression"
"Small interfering RNA (siRNA)-mediated depletion of DNMT3A and DNMT3B specifically increased PLAKOGLOBIN gene promoter activity and expression, and subsequently abrogated autocrine hGH stimulated mammary carcinoma cell migration"<-DNMT3A/B and therefore GH may reduce migration and encourage mesenchymal condensation.
HGH represses PLAKOGLOBLIN promoter activity.
Different tissue than chondrocytes but I think we can safely assume that HGH increases DNA methylation. However, we don't know if HGH affects telomere length and both DNA methylation and telomere lengthening are needed to avert cellular senescence.
Growth hormone mediates pubertal skeletal development independent of hepatic IGF-1 production.
"Deficiencies in either growth hormone (GH) or insulin-like growth factor 1 (IGF-1) are associated with reductions in bone size during growth in humans and animal models. Liver-specific IGF-1-deficient (LID) mice, which have 75% reductions in serum IGF-1, were created previously to separate the effects of endocrine (serum) IGF-1 from autocrine/paracrine IGF-1. However, LID mice also have two- to threefold increases in GH, and this may contribute to the observed pubertal skeletal phenotype. To clarify the role of GH in skeletal development under conditions of significantly reduced serum IGF-1 levels (but normal tissue IGF-1 levels), we studied the skeletal response of male LID and control mice to GH inhibition by pegvisomant from 4 to 8 weeks of age. Treatment of LID mice with pegvisomant resulted in significant reductions in body weight, femur length (Le), and femur total area (Tt.Ar), as well as further reductions in serum IGF-1 levels by 8 weeks of age, compared with the mean values of vehicle-treated LID mice. Reductions in both Tt.Ar and Le were proportional after treatment with pegvisomant. On the other hand, the relative amount of cortical tissue formed (RCA) in LID mice treated with pegvisomant was significantly less than that in both vehicle-treated LID and control mice, indicating that antagonizing GH action, either directly (through GH receptor signaling inhibition) or indirectly (through further reductions in serum/tissue IGF-1 levels), results in disproportionate reductions in the amount of cortical bone formed. This resulted in bones with significantly reduced mechanical properties (femoral whole-bone stiffness and work to failure were markedly decreased), suggesting that compensatory increases of GH in states of IGF-1 deficiency (LID mice) act to protect against a severe inhibition of bone modeling during growth, which otherwise would result in bones that are too weak for normal and/or extreme loading conditions."
"growth hormone receptor knockout (GHRKO) mice have decreased femoral length,"
"GH stimulates IGF-1 production"
"the reduction in mean femoral length (Le) owing to pegvisomant was not significantly different between LID (approximately 7%) and control mice (approximately 5%). "
"Ghr/Igf1 nullizygotes have more severe growth retardation than mice with single ablations of either the Ghr or the Igf1 gene."
"Femur length (Le) was reduced significantly in pegvisomant-treated LID mice, indicating reduced GH action in the femoral growth plate. Interestingly, these reductions in femur length (Le) were proportionally matched to reductions in transverse bone size such that pegvisomant-treated LID mice (as well as control mice) had identical robustness values when compared with vehicle-treated animals. Thus loss of GH action during puberty appears to hinder both transverse and longitudinal bone growth in a coordinated fashion, whereas loss of hepatic IGF-1, as reported previously in the LID mouse, reduces transverse bone growth but not longitudinal growth, thereby resulting in a less robust, more slender phenotype for LID mice"
I mentioned in an earlier blog entry about how bone density scans(Dual energy X-ray absorptiometry) also detect changes in size. Researchers define volumetric bone mineral density as the mass divided by volume(we of course are striving to increase the volume, so an increase in bone size can actually lead to the same levels of vBMD if both mass and volume increase at the same rate). vBMD can be useful for our purposes as a sign of trabecular microfractures because if you don't cause trabecular microfractures you're not going to cause microfractures in the cortical bone.
"Male runners present generally higher BMD than sedentary individuals. We postulated that the proximal tibia BMD is related to the running distance as well as to the magnitude of the shocks (while running) in male runners."
Running at minimum causes trabecular microfractures and increases bone mineral content. The theory was that the amount of microfractures caused is related to the force of impact generated while running and the duration of exercise.
"Several measurements were performed at the proximal tibia level: volumetric BMD (vBMD), cortical index (CI) i.e. an index of cortical bone thickness and peak accelerations (an index of shocks during heel strike) while running (measured by a 3-D accelerometer)."
The cortical index could be very useful to us as a measurement tool to determine when cortical microfractures have occurred.
"CI and vBMD a) increase with running distance to reach a plateau over 30 km/wk, b) are positively associated with peak accelerations over 30 km/week."
"CI and vBMD are associated with the magnitude of the shocks during heel strike in runners."
The faster you run and the harder you strike the ground the more microfractures you cause. The fact that cortical index leveled off over time goes against our microfracture theory. As you should always be able to cause microfractures(unless there's a performance issue where you're not able to run as hard or fast or there's an adaptation issue where over time the same impact no longer causes cortical microfractures). Perhaps the cause of the increase in cortical bone size is by other factors than microfracture healing.
Note however that the sedentary individuals are actually the tallest and that the more the individuals ran per week the shorter they tended to be. This could be due to proteoglycan depletion and other factors from articular cartilage. It could also be due to shorter individuals enjoying going running more. There would have to be a longitudinal study done to know the true effect of running on height.
Note in this study the control group also tended to be taller than the runners but there was seemingly no correlation between height and distance ran like in the other study.
"As hypothesised, tibia diaphyseal bone mineral content (vBMC), cortical area and polar moment of resistance were largest in sprinters, followed in descending order by middle and long distance runners, race-walkers and controls. When compared to control people, the differences in these measures were always >13% in male and >23% in female sprinters (p<0.001)."
Diaphysis is the midshaft section of the bone. The healing of microfractures induced by running would increase bone mineral content in that area. The use of the terminology cortical area rather than cortical volume alludes to the fact their was no increase in height.
"Similarly, the periosteal circumference in the tibia shaft was larger in male and female sprinters by 4% and 8%, respectively, compared to controls (p<0.001)."
Sprinting creates a strong enough chemical and mechanical signaling to increase periosteal width. Periosteal width is different from the deposition of new bone beneath the periosteum.
"Epiphyseal group differences were predominantly found for trabecular vBMC in both male and female sprinters, who had 15% and 18% larger values, respectively, than controls (p<0.001)."
So trabecular microfractures were caused first, followed by cortical microfractures, and followed by an increase in periosteal width.
This study did not take place over a duration of time so it could not have studied a change in bone length.
What we learned though is that sprinting is sufficient to cause cortical microfractures by the fact that cortical area increased. There's no basis as to why if cortical length and width can increase that cortical height can't increase as well(unless sprinting causes microfractures in a way that only results in an increase of length or width of the bone when the microfractures are healed). However, there doesn't tend to be cortical bone in the epiphysis. The reason why periosteal width can increase with some stimulus and the epiphysis can't is that they are different tissues. To increase the height of the epiphysis requires special stimulation. The healing mechanisms for bone differ from how the cortical bone is aligned as cortical bone tends to increase in width rather than height(likely due to the OPG/RANKL gradient).
Note that the long distance runners had the greatest epiphyseal area. This could however be due to less modeling into cortical bone.
Growth and adaptation to mechanical loading of immature bone and articular cartilage
"The osteogenic response due to different running modes was greatest in the femoral metaphysis. In particular, solely downhill running with high proportions of eccentric loadings was able to increase femoral trabecular bone mineral density in the metaphysis, while level running with similar amounts of concentric and eccentric loadings failed to induce an osteogenic response in this area. In contrast, neither in the dia- nor in the epiphysis any running mode-related structural alteration could be found. Moreover, downhill running increased femoral cartilage height and COMP staining height in a site-specific manner, while level running was insufficient to induce such alterations. These changes were not accompanied by any signs of degeneration. In contrast, cartilage thickness, mechanical properties, and expression of major cartilage network proteins (i.e., collagen II, collagen IX, COMP, and matrilin-3) in tibial cartilage remained unaffected by the different running modes. Growth was associated with high modeling of the morphology, biochemical composition, mechanical properties of articular cartilage, and structure of subchondral bone. COMP showed a profound redistribution throughout the cartilage. Moreover, growth diminished cartilage thickness, distribution and amounts of the matrix proteins collagen II, collagen IX, and matrilin-3 and related to this decreased its compressive properties. Functional condensation of the subchondral trabecular bone and subchondral plate due to enchondral ossification was also found to occur with increasing age, which did not seem to affect cartilage mechanical properties. In conclusion, these are the first in vivo data illustrating the effect of different running modes with different proportions of eccentric and concentric loadings on immature bone and articular cartilage in such a physiological and systematic manner."
This study was in a foreign language and I could not get the full thesis.
HEre's the running study though:
Effect of different running modes on the morphological, biochemical, and mechanical properties of articular cartilage
"Mechanical loading plays an important role not solely in cartilage development, but also in cartilage degeneration. Its adaptation behavior to mechanical loading has not been clearly delineated. The aim of the study was to examine the effect of different running modes (with different muscle contraction types) on morphological, biochemical, and mechanical properties of articular cartilage in the knee of growing rats. Thirty-six female Sprague-Dawley rats were randomly assigned into a nonactive age-matched control (AMC), level (LEVEL), and 20° downhill (DOWN) running group (n = 12 each). Running groups were trained on a treadmill for 30 min/day, 5 days/week for 6 weeks. Immunohistochemical staining and analysis of expression for collagen II, collagen IX, cartilage oligomeric matrix protein (COMP), and matrilin-3, histomorphometry of femoral cartilage height and femoral COMP staining height, and indentation testing of tibial articular cartilage were performed. Rats subjected to downhill running showed a significantly (P = 0.015) higher COMP staining height and a tendentially (P = 0.084) higher cartilage height in the high-weight bearing area of femoral articular cartilage. Cartilage thickness, mechanical properties, and expression of cartilage network proteins in tibial cartilage remained unaffected by different running modes. Our data suggest that joint loading induced by eccentric muscle contractions during downhill running may lead to a site-specific adaptation."
couldn't get full study
Here's a study linking muscular contractions to articular cartilage deformation.
There is actually mixed data on whether running is anabolic:
The influence of cyclic concentric and eccentric submaximal muscle loading on cell viability in the rabbit knee joint
"Cartilage loading is associated with the onset and progression of osteoarthritis and cell death may play an important role in these processes. Although much is known about cell death in joint impact loading, there is no information on joints loaded by muscular contractions. The aim of this study was to evaluate the influence of muscle generated eccentric and concentric submaximal joint loading on chondrocyte viability. We hypothesised that eccentric muscle activation leads to increased cell death rates compared to concentric loading and to controls. 16 rabbits received either 50 min of uni-lateral, cyclic eccentric (n=8) or concentric (n=8) knee loading. Muscle activation for these dynamic conditions was equivalent to an activation level that produced 20% of maximum isometric force. Contralateral joints served as unloaded controls. Cell viability was assessed using confocal microscopy. Eccentric contractions produced greater knee loading than concentric contractions. Sub-maximal contractions caused a significant increase in cell death in the loaded knees compared to the unloaded controls, and eccentric loading caused significantly more cell death than concentric loading. Cyclic sub-maximal muscle loading of the knee caused increased chondrocyte death in rabbits. These findings suggest that low levels of joint loading for prolonged periods, as occurs in endurance exercise or physical labour, may cause chondrocyte death, thereby predisposing joints to degeneration."
It's unclear why this study got a catabolic result and the other one didn't. They did measure different things. Couldn't get full study.
"Mice were divided into five groups: sedentary (SED), uphill training (TRU), downhill training (TRD), excessive uphill training (ETU), and excessive downhill training (ETD) for an 8-week training intervention protocol. ETD group had increased pro-inflammatory cytokines in serum, vastus lateralis (VL), and vastus medialis (VM) muscles, while ETU group mice had increased cytokine levels in the VL and VM. Total knee joint OARSI score were more significant in ETD group compared to SED and TRU groups. They were also more meaningful for the medial tibial plateau of ETD group compared to SED group. MMP-3 and cleaved Caspase-3 were higher in the ETD group than the SED and TRU group, while Adamts-5 was higher in the ETD group than the SED group. TRU group had increased PRG-4 levels compared to ETU and ETD group. ETD group had decreased total bone volume, trabecular bone volume, and cortical thickness compared to SED group. Excessive downhill training induced a chronic pro-inflammatory state in mice and was associated with early signs of cartilage and bone degeneration that are clinical indicators of knee OA."
Key term is excessive.
"The moderate uphill and downhill training protocols lasted for 8 weeks, and the animals ran 3 days a week (Mon/Wed/Fri) for a total of 1080 min of running. The uphill and downhill overtraining protocols consisted of five consecutive days of training (Mon–Fri) for a total volume of 2475 min"
"Increased levels of IL-1beta have been observed in OA cartilage, and high serum levels of IL-6 and TNF-alpha have been associated with cartilage loss. Moreover, IL-1beta promotes the release of MMP-3 (Matrix Metalloproteinase-3) and the aggrecanase Adamts-5 (a disintegrin and metalloproteinase with thrombospondin motifs-5), enzymes involved in articular cartilage matrix degradation, a hallmark of OA. Also, metabolic inflammation has been related to increased chondrocyte apoptosis. In contrast, PRG-4 (Proteoglycan 4/Lubricin) reduces friction between biological surfaces promoting tissue homeostasis"
Here's the articular cartilage:
Uphill and downhill training looks like it has thicker cartilage than controls but I can't tell.
"High joint loads can trigger bone remodeling"
Long-term repetitive mechanical loading of the knee joint by in vivo muscle stimulation accelerates cartilage degeneration and increases chondrocyte death in a rabbit model
"24 rabbits were divided into an (i) eccentric, (ii) concentric, or (iii) isometric knee extensor contraction group (50 min of cyclic, submaximal stimulation 3 times/week for four weeks=19,500 cycles) controlled by the stimulation of a femoral nerve cuff electrode on the right hind limb. The contralateral knee was used as a non-loaded control. The knee articular cartilages were analysed by confocal microscopy for chondrocyte death, and histologically for Mankin Score, cartilage thickness and cell density. All loaded knees had significantly increased cell death rates and Mankin Scores compared to the non-loaded joints. Cartilage thicknesses did not systematically differ between loaded and control joints. Chondrocyte death and Mankin Scores were significantly increased in the loaded joints, thereby linking muscular exercise of physiologic magnitude but excessive intensity to cartilage degeneration and cell death in the rabbit knee."
couldn't get full study. Don't know why this was found to be catabolic.
In Vivo Dynamic Deformation of Articular Cartilage in Intact Joints Loaded by Controlled Muscular Contractions
"When synovial joints are loaded, the articular cartilage and the cells residing in it deform. Cartilage deformation has been related to structural tissue damage, and cell deformation has been associated with cell signalling and corresponding anabolic and catabolic responses. Despite the acknowledged importance of cartilage and cell deformation, there are no dynamic data on these measures from joints of live animals using muscular load application. Research in this area has typically been done using confined and unconfined loading configurations and indentation testing. These loading conditions can be well controlled and allow for accurate measurements of cartilage and cell deformations, but they have little to do with the contact mechanics occurring in a joint where non-congruent cartilage surfaces with different material and functional properties are pressed against each other by muscular forces. The aim of this study was to measure in vivo, real time articular cartilage deformations for precisely controlled static and dynamic muscular loading conditions in the knees of mice. Fifty and 80% of the maximal knee extensor muscular force (equivalent to approximately 0.4N and 0.6N) produced average peak articular cartilage strains of 10.5±1.0% and 18.3±1.3% (Mean ± SD), respectively, during 8s contractions. A sequence of 15 repeat, isometric muscular contractions (0.5s on, 3.5s off) of 50% and 80% of maximal muscular force produced cartilage strains of 3.0±1.1% and 9.6±1.5% (Mean ± SD) on the femoral condyles of the mouse knee. Cartilage thickness recovery following mechanical compression was highly viscoelastic and took almost 50s following force removal in the static tests"
"Cartilage deformation is known to cause deformations of the chondrocytes and their nuclei, and these deformations, in turn, are known to affect the biological signaling response of chondrocytes that control the maintenance and adaptation of the tissue"
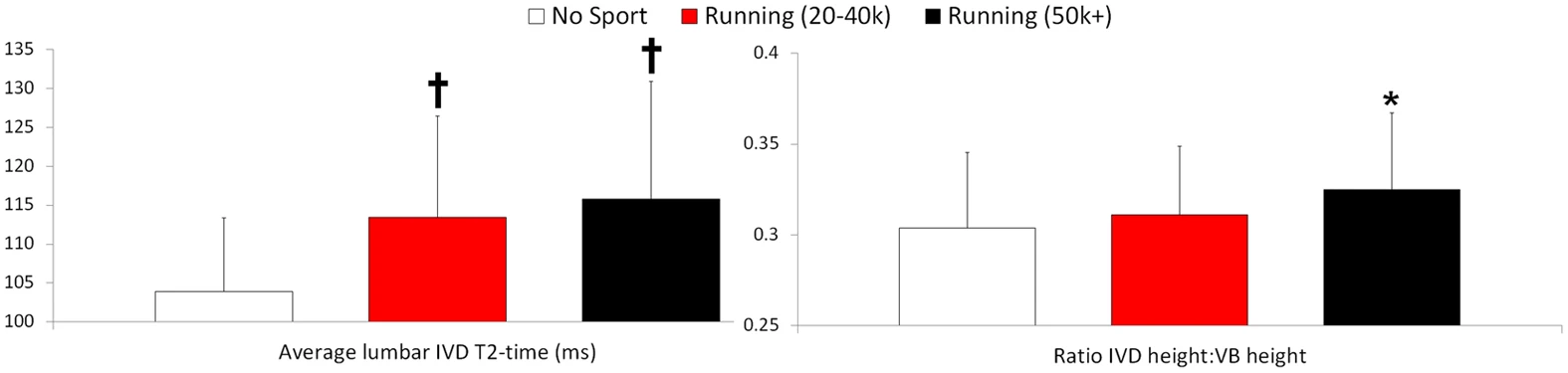
"There is currently no evidence that the intervertebral discs (IVDs) can respond positively to exercise in humans. Some authors have argued that IVD metabolism in humans is too slow to respond anabolically to exercise within the human lifespan. Here we show that chronic running exercise in men and women is associated with better IVD composition (hydration and proteoglycan content) and with IVD hypertrophy{IVD Hypertrophy is possible height}. Via quantitative assessment of physical activity we further find that accelerations at fast walking and slow running (2 m/s), but not high-impact tasks, lower intensity walking or static positions, correlated to positive IVD characteristics. These findings represent the first evidence in humans that exercise can be beneficial for the IVD and provide support for the notion that specific exercise protocols may improve IVD material properties in the spine."
"“likely anabolic loading window” for the IVD exists: dynamic loading of 0.2–0.8 MPa, generating intra-discal pressures of approximately 0.3–1.2 MPa, at 0.1 to 1 Hz for approximately eight hours a day."
"no sport (referents), 20–40 km per week running (joggers), or 50 + km per week running (long-distance runners)."
"The height of the IVD relative to that of the vertebral body, an indicator of IVD hypertrophy, was greater in the long-distance runners"
" Runners have more hydrated and hypertrophied lumbar IVDs."
This is pretty significant. "Higher T2-times indicate better IVD hydration and glycosaminoglycan content."
Zydrunas Savickas is the athlete pictured above. You can see a bruise on his left forearm. Perhaps a hematomma from a microfracture. I couldn't find a good image of Zydrunas Savickas in stationary mode to compare his bone length in performing the farmers walk versus stationary. If the loads that a multi-time strongman champion are not enough to provide a measurable increase in bone length then that does not bode well for the rest of us.
Strongman is the sport with the greatest potential to cause microfractures. Exercises like the shield carry, farmer's walk, and the refrigerator carry are high impact, high load exercises that have the potential to cause cortical bone microfractures.
The extremely large muscles of most strongmen competitors makes it hard tell exactly how their bones have adapted to the stress placed upon them. That's exactly the problem with the mechanostat theory at present.
The microstrain graph is vague. Bone Density can increase in the following ways: size, BMC, and porosity. Bone Mineral Content can be induced by chemical means so it's logical to assume that it's threshold to induce that method of adaptation is lower. Periosteal width is not directly inhibited such as how growth in the long bones are inhibited so at some amount of chemical and mechanical theory periosteal growth is induced.
Trabecular microfractures occur before cortical microfractures. The definition of the fracture point by a mechanostat model is when maximum deformation is exceeded. Cortical microfractures should occur before then as when you're breaking a pencil when you bend their is a microscopic denaturing(change in the pencils shape) before the pencil finally snaps. So here's what needs to be added to the mechanostat model:
At 1500 microstrain is probably where Bone Mineral Content begins to increase as that is the easiest form of bone adaptation.
Other points needed on the graph:
X1: Point where Trabecular microfractures begin to occur
X2: Point where periosteal width increases(although the periosteum is not likely to increase in width as a result of the pulling apart of bones it is possibly that the periosteum will increase in width as a result of impact which does temporarily change the length of the bone)
X3: Point where Cortical Microfractures occur
What good is knowing the bone modeling range if we don't receive data on when a specific form of bone modeling(cortical microfracture healing) to increase height occurs?
This article is incredibly outdated right now and I don't know how to fix it as of yet.
The mechanostat theory is problematic to theory of creating microfractures in the cortical bone to increase height. The mechanostat theory essentially states that it takes a 1.5% change in bone length to cause a fracture in the cortical bone. The Mechanostat theory does not distinguish between fracture and microfracture.
The mechanostat theory states that bone modeling is incited when the bone changes in length by .15%. It's not clear whether the adaptation is a result of a change in the width of the periosteum or is it a case of trabecular microfracture.
It is more likely to be a healing of trabecular microfractures thus resulting in decreased porosity of the bone. The three determinants of bone density being size(what we want), porosity, and bone mineral content. It's pretty clear that bone modeling is possible with average weights and exercise. If the bone modeling range is achieved by say 20lbs dumbell bench which could compress the bone by .15%, then it would take a 200lbs dumbell bench to compress the bone by 1.5%. Using 200lbs dumbells on the bench is elite level.
So, we either have to find exercises that change the bone length by 1.5% or we have to prove that the mechanostat is flawed. Given that the mechanostat theory was developed in 1960 and that bone research remains heavily flawed even today, I choose the latter option. It's hard to believe that all the bone microfractures would occur in the trabecular bone and none would occur in the cortical bone in the bone modeling range.
The Utah paradigm of skeletal physiology: an overview of its insights for bone, cartilage and collagenous tissue organs.
"...adding them to the 1960 paradigm led to the still-evolving Utah paradigm of skeletal physiology that concerns, in part, how load-bearing skeletal organs adapt to the voluntary mechanical loads on them."
So it is possible that cortical microfractures occur in the bone modeling range and therefore it is possible to perform plausible exercises to cause them. Crisis averted.
We have to encourage scientists to look less at stress-strain laws and the growth of bone and more on microfractures.
I thought this article had some instructional purposes even though it dealt with bone remodeling. Bone modeling some say may be one possible way to increase height(such as surface osteoblasts depositing new bone on the subchondral plate) but bone modeling can occur in ways that do not increase height such as a decrease in porosity or an increase in width of the periosteum(an increase in periosteal width can increase height if it is an increase in width on the flat bone of the skull). Bone remodeling is mostly rejuvenating old bone with new bone whereas bone modeling changes the structure and shape of the bone.
Even though the article deals with bone remodeling it does mention some facts about microfracture healing.
Mathematical modeling of spatio-temporal dynamics of a single bone multicellular unit.
"During bone remodeling, bone-resorbing osteoclasts and bone-forming osteoblasts are organized in bone multicellular units (BMUs), which travel at a rate of 20-40 mum/d for 6-12 mo, maintaining a cylindrical structure. However, the interplay of local BMU geometry with biochemical regulation is poorly understood."
A bone multicellular unit is also known as an osteon.
"We developed a mathematical model of BMU describing changes in time and space of the concentrations of proresorptive cytokine RANKL and its inhibitor osteoprotegerin (OPG), in osteoclast and osteoblast numbers, and in bone mass. We assumed that osteocytes surrounding a microfracture produce RANKL, which attracted osteoclasts"
Osteoclasts are essential for proper bone remodeling and bone growth.
"OPG and RANKL were produced by osteoblasts and diffused through bone, RANKL was eliminated by binding to OPG and RANK. Osteoblasts were coupled to osteoclasts through paracrine factors. The evolution of the BMU arising from this model was studied using numerical simulations. Our model recapitulated the spatio-temporal dynamics observed in vivo in a cross-section of bone. In response to a RANKL field, osteoclasts moved as a well-confined cutting cone. The coupling of osteoclasts to osteoblasts allowed for sufficient recruitment of osteoblasts to the resorbed surfaces. The RANKL field was the highest at the microfracture in front of the BMU[more osteoclasts were attracted to the front of the osteon, by front what's likely meant is away from the center of the bone], whereas the OPG field peaked at the back of the BMU[so more osteoblasts tended to propagate torwards the center of the bone], resulting in the formation of a RANKL/OPG gradient, which strongly affected the rate of BMU progression and its size."
Manipulating RANKL and OPG fields may eventually be a way to grow taller. You could for example put two OPG fields at the top and bottom of the bone to grow taller that way.
A comparison of the fatigue behavior of human trabecular and cortical bone tissue.
"The results showed that trabecular specimens had significantly lower moduli and lower fatigue strength than cortical specimens, despite their higher mineral density values. Fracture surface and microdamage analyses illustrated different fracture and damage patterns between trabecular and cortical bone tissue, depending upon their microstructural characteristics."
It's easier to cause microfractures in cortical bone than trabecular bone.
Causing microfractures in the cortical bone is not quite related to the mechanostat theory of bone adaptation(the theory provides a model of how bone adapts to strain). But the mechanstat theory provides us with a model of how much strain is needed to cause a fracture which is about 15000 units of strain. 1000 Units of Strain is a 0.1% change in length of the bone. So if you were in a medievial rack you would need your bones to be pulled apart by 1.5% to cause a fracture.
According to wikipedia, "The fracture load for axial forces of the Tibia for example is about 50 to 60 times the body weight."
Axial means along the transverse plane.
"The fracture load for forces perpendicular to the axial direction is about 10 times lower"
That load is about 5 to 6 times bodyweight which is still a lot more than people can lift(except in the leg press).
The load needed to cause cortical microfractures is unclear.
A possible experiment would be loaded hangs. Measure the original length of the radius(a forearm bone). And continue to hang with more attached weight until the length of the raidus increases by 1%. Then perform an X-ray for microfracture.
Not only possible but viable as well. Limb lengthening works but at the expense of being able to perform well athletically and at great monetary cost.
Our theory is that it is possible to grow taller via microfractures sufficient enough to denature the bone to the point where their is room for new bone to grow(and thus make the body taller) but not so large as to the point to denature the bone where it can not function as well as it once did(like limb lengthening surgery). It's also necessary to induce these microfractures while under tensile strain(stretching the bone) as microfractures induced under tensile strain are different than other kinds of microfractures.
The following types of articles can be used to support this theory:
1) Articles that suggest that microfractures denature the bone - We found this.
2) Articles that suggest that the bone can grow wider by means other than the periosteum(the periosteum is a width specific mechanism as articular cartilage is what's present at the end of the bone) Bones can grow wider by means other than the periosteum by things like surface osteoblasts and direct secretion by osteocytes.
3) Any article that explains the physiology of how microfractures are healed.
4) The histology of microfractures. We could always use more on this as histology would let us know exactly how bone works which would be applicable to other height increase methods as well.
The following types of articles can be used to prove that the theory is viable and able to be put into practice:
1) Articles that show activities that cause microcracks in the bone and these activities are such that they can be performed in a gym environment or with no outside materials at all. Again not just any microcracks but tensile strain microcracks. Which exercises can induce these?
Here's an article about trabecular microfracture versus cortical microfracture:
Trabecular microfracture precedes cortical shell failure in the rat caudal vertebra under cyclic overloading.
"In the current study we determine the relationship between applied cyclic compressive overloading and the resulting amount of microdamage in isolated rat tail vertebrae, a bone that has been used previously for in vivo loading experiments[compressive overloading may not be the same as tensile strain overloading but some of the principles may apply]. Rat caudal vertebrae (C7-C9, n = 22) were potted in bone cement and subjected to cyclic compressive loading from 0 to 260 N."
Netwon's are kg*m/s^2. In vivo simply means that the experiment was performed on live rats. Rats tend to weigh about 0.5kg. So the compressive loading was quite a bit relative to a rat's size.
"In cancellous bone, trabecular microfracture was the primary form of microdamage observed with few microcracks. Trabecular microfracture prevalence increased with the amount of cyclic loading and occurred in nine out of 10 specimens loaded into the tertiary phase[The more you load the more likely you are to experience trabecular microfracture, although we don't know if trabecular microfracture could help with LSJL, it would probably reduce hydrostatic pressure but could allow for easier flow of bone marrow]. Only small amounts of microdamage were observed in the cortical shell of the vertebrae, demonstrating that, under axial cyclic loading, damage occurs primarily in regions of cancellous bone before overt fracture of the bone (macroscopic cracks in the cortical shell)."
Trabecular bone is synonymous with spongy bone which is not the kind of microfracture damage we want if we want to grow taller by tensile strain(it may be helpful for other means). However, it does state that microfractures occurred in the cortical(compact) bone.
The study used axial loading but if you tilted the vertebrae on it's side for example and used compressive loading that way you be able to get tensile strain in the direction you want. Again though it takes much more to cause sufficient strain to cause microfractures then it does to generate sufficient hydrostatic pressure.
The body doesn't naturally want to increase height via bone size. What the body wants to do is grow outward via the periosteum(articular cartilage is at the ends of bones) or decrease the porosity of the bone thus increasing bone density. You can also see that their are a lot of blood vessels and that blasted Haversian canal getting in our way of height growth.
But most of the impact, spiral, compression, and strain forces on bone are absorbed by the compact bone rather than the spongy bone. Building stronger bones by exercise without microfractures will only increase bone growth outward via the periosteum and increased bone mineral content, microfractures must be caused to induce height growth. We must make sure the forces on the compact bone are strong enough to cause them.
Causing microfractures in the compact bone is sufficient as "if for any reason there is an alteration in the strain to which the cancellous subjected there is a rearrangement of the trabeculae"(again wikipedia). Thus, if you make your compact bone taller the internal spongy bone will adapt as well. Via microfractures we are causing microscopic seperations in the bone and allowing room for new bone tissue to be laid down.
In an earlier entry, I mentioned impact pushups(pushing your body off the ground in push up position and then landing using your hands causing impact) and sprints as ways to cause microfractures without weights.
Here are some microfracture inducing exercises that include weights:
Heavy squat walkouts with stomping to increase the impact(sort of like the strongman event the refrigerator carry except you are stomping as hard as you can in one place)
Hanging while twisting with a heavy dip belt(the twisting provides spiral forces on the bone)
Decline bench hold, holding dumbells rather than plates(dumbells are easier to hold than plates. This exercise causes microstrain on the bone)
Impacting all your bones with weights or a hammer(the heavier the weight you use the less force you can exert with it. We want a weight that maximizes the equation F=ma)
The most important exercise is the heavy squat walkouts with stomps which will cause heavy microfractures in the femur/tibia/spine. Where the bar is in the squat is the optimal location for holding the heaviest amount of weight and inducing the most amount of impact throughout the entire body.
Don't believe that stuff about deadlifts and squats increasing GH and therefore increasing height. Again, that will only make the bones wider via the periosteum. Microfractures are the way to go :)